Executive summary
The White House’s recent Executive Order on AI addresses risks from AI applied to biology. Developers training machine learning models on primarily biological sequence data using more than 1e23 operations are required by the Executive Order to report to the federal government about their cybersecurity, red-teaming, and other risk management efforts made during the development of these models.
This report provides an overview of our newly curated dataset focused on biological sequence models. Our dataset contains comprehensive information on nearly a hundred biological sequence models, including the specific datasets used for their training, their intended tasks, and the availability of the model weights. Additionally, our analysis covers 30 biological sequence datasets, collectively containing billions of protein sequences. Our focus in this analysis includes:
The training compute trends of biological sequence models
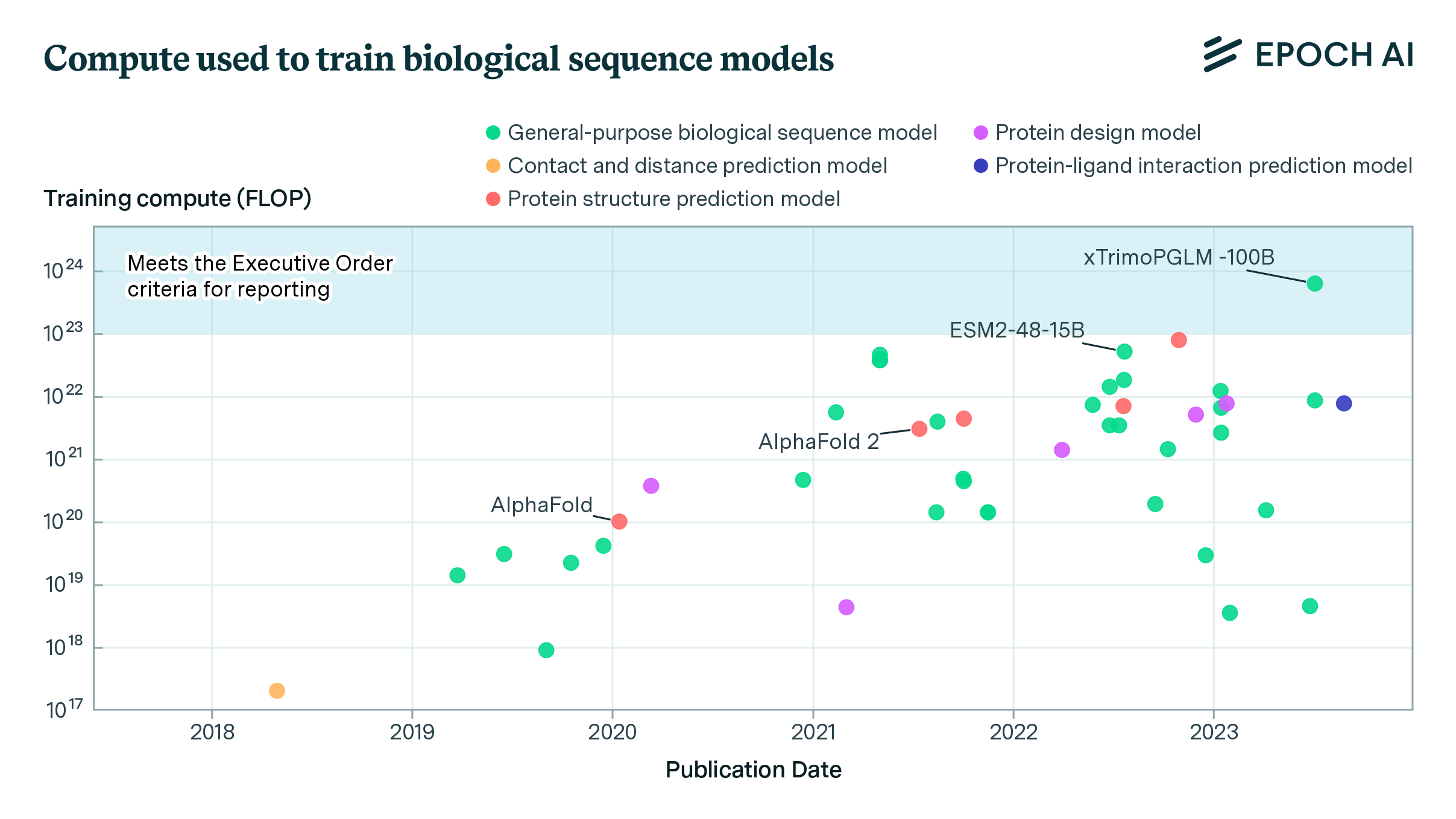
- xTrimoPGLM-100B (Chen et al., 2023), a 100B-parameter protein language model, exceeds the Executive Order’s reporting threshold of 1e23 operations by a factor of six. Over a dozen models are within a factor of 10x of the Executive Order’s reporting threshold.
- The amount of compute used to train biological sequence models has, over the past 6 years, increased by an average of 8-10 times per year. This rapid growth in training compute outpaces the rate of growth for computer vision and reinforcement learning systems, but is comparable to the growth rate for language models.1
Biological sequence data
- There are extensive public databases providing billions of protein sequences and structural data entries. Key protein sequence databases include UniRef (365M sequences), UniProtKB (252M), MGnify (3B), BFD (2.5B), and MetaClust (1.6B).
- The total stock of protein sequence data across key databases is estimated to be approximately 7B entries, an order of magnitude greater than the amount used so far for training state-of-the-art biological sequence models today.
- The total stock of protein and DNA sequence data is rapidly expanding, with key databases growing at rates of 31% for DNA sequences, 20% for metagenomic protein sequences, and 6.5% for protein structures between 2022 and 2023.
- The majority of protein sequences are derived from translating known DNA/RNA sequences. In addition, experimentally verified protein structures from PDB and AI-predicted structures from AlphaFoldDB and ESMAtlas provide a rapidly expanding complementary resource for model training.
This analysis provides a comprehensive overview of biological sequence models, including the compute resources and training data used. It allows extrapolation of future compute growth trends, providing valuable insights into which models when scaled further will likely require regulatory scrutiny. Our work further highlights a potential regulatory gap in the current formulation of the training compute threshold for regulatory oversight. Specifically, the Executive Order suggests that AI models that incorporate significant but not majority biological data may evade stringent oversight despite using more than 1e23 operations during training. This could potentially permit the training of models on extensive biological sequences without being subject to heightened reporting requirements.
The AI Executive Order
The Executive Order issued in October 2023 by the White House puts forth a comprehensive set of measures to address potential national security risks from the development and deployment of artificial intelligence (AI) systems. This order requires companies developing “dual-use foundation models” to submit regular reports to the government. These reports should detail their model development plans and risk management strategies, including efforts to safeguard model weights and red-team test models to identify dangerous capabilities.
The Executive Order states that the Secretary of Commerce will determine and update the definition of a “dual-use foundation model” subject to reporting requirements. However, it outlines two specific categories of models that fall under reporting in the interim before the Secretary issues an official definition:
[A]ny model that was trained using a quantity of computing power greater than 10^26 integer or floating-point operations, or using primarily biological sequence data and using a quantity of computing power greater than 10^23 integer or floating-point operations.
Entities developing AI models trained on “primarily biological sequence data” using more than 1e23 operations face new reporting and compliance requirements. In addition to models trained on biological data, the Order expands oversight to encompass large foundation models that could potentially be misused, despite not working exclusively with biological sequences. Typically containing tens of billions of parameters, these dual-use models warrant scrutiny because their scale and versatility makes them ripe for exploitation if misapplied to high-risk domains.
The order provides a broad directive but does not specify the standards for the required reports, the exact nature of the red-team tests, and the cybersecurity measures that labs must implement. The task of filling in any gaps is delegated to the National Institute of Standards and Technology (NIST). NIST is to work in coordination with the Secretary of Energy and the Secretary of Homeland Security to develop frameworks for risk evaluations and regulation later this year. These frameworks will guide the training, deployment, dissemination, and application of AI models that work with biological sequence data, ensuring their safe and responsible use.
Epoch AI has previously published information on many large-scale machine learning models (Epoch AI, 2024). However, this order sets a lower reporting threshold of 1e23 operations specifically for biological sequence models. By focusing on models impacted by the 1e23 threshold, this analysis spotlights those likely needing regulatory disclosure under the order’s heightened governance for AI applied to biology.
Models trained on biological sequence data
In the wake of AlphaFold 2’s transformative performance at the Critical Assessment of Structure Prediction (CASP) 14 competition in 2020, there has been rapidly growing interest in applying machine learning methods to problems in biology (Read et al., 2023). The majority of biological sequence models are trained on protein (amino acid), DNA or RNA sequences. Biological sequence models are trained to perform various tasks. Key tasks include:
- Protein structure prediction: AlphaFold 2 (Jumper et al., 2021), RoseTTAFold (Krishna, 2023), ESMFold (Lin et al., 2022) are prominent examples in this group. These models predict the three-dimensional structure of proteins directly from amino acid sequences.
- Protein design: Fold2Seq (Cao et al., 2021), ProGen (Madani et al. 2023) and ProteinGAN (Repecka et al., 2021) are members of this group. These models are designed to generate novel protein sequences that adhere to specific structural or functional constraints. They are employed in the field of synthetic biology for designing proteins with desired characteristics. Some models such as ZymCTRL (Munsamy et al., 2022) are specialized to generate proteins of a specific class, such as enzymes.
- Contact and distance prediction: Models such as DMPfold (Greener, 2019) and DeepConPred2 (Ding, 2018) exemplify this group. These models specialize in predicting the distances and contacts between amino acid residues within a protein. Such predictions are crucial for understanding protein folding mechanisms and for improving the accuracy of structure prediction models.
- Protein-ligand interaction prediction: This category encompasses models that are important for predicting binding behavior between specific molecules and proteins. Examples include LEP-AD (Daga et al. 2023), which predicts protein-ligand binding affinity and BERT-RBP (Yamada and Hamada, 2022), which predicts interactions between RNA sequences and RNA-binding proteins.
- Gene regulatory element prediction: Models trained on genetic sequences such as HyenaDNA (Nguyen et al. 2023) and DNABert (Ji et al. 2021) are trained to predict regulatory elements such as promoters, enhancers and splice sites within DNA sequences.
- General-purpose biological sequence models: General-purpose biological sequence models are models that learn representations of biological sequences in a task-agnostic manner. They are trained on large datasets of unlabeled protein, DNA, or RNA sequences to capture structural and functional properties common across diverse biomolecules. Examples include Ankh (Elnaggar et al., 2023) and xTrimoPGLM-100B (Chen et al., 2023). Fine-tuning allows general-purpose models to be harnessed to predict functional effects of sequence variation, protein subcellular localization, protein structures, protein interactions and other aspects of protein function.
These distinctions are not rigid boundaries, as models developed for one task can often be repurposed for others. For example, AlphaFold 2 was originally designed for protein structure prediction, but it has been successfully adapted for many other applications, including predicting whether any single-amino acid change in the human proteome leads to disrupted protein function (AlphaMissense, Cheng et al. 2023) and predicting multi-chain protein complexes (AlphaFold-Multimer, Evans et al. 2021).2
Biological sequence models often use architectures which are common in natural language processing, such as LSTMs and, notably, the transformer (Hu et al., 2022). For example, the Transformer architecture (Vaswani et al., 2017) used in leading language models such as OpenAI’s GPT and Google DeepMind’s Gemini has been employed for biological sequence models such as Nucleotide Transformer (Dalla-Torre et al., 2023) and xTrimoPGLM-100B (Chen et al., 2023).
When architectures from natural language processing are applied to biological sequences, the resulting models are often called protein language models (pLMs) in the case of protein sequences or nucleotide language models (nLMs) in the case of DNA or RNA sequences. Like other foundation models (Bommasani et al., 2021), pLMs and nLMs are trained with large volumes of compute and unlabeled data, and can be adapted for a variety of downstream tasks. Many state-of-the-art protein structure prediction models (such as ESMFold, OmegaFold) are adapted protein language models (see Hu et al., 2022 for a survey of pLMs for structure prediction). pLMs have benefited greatly from scaling up model size and compute (Chen et al., 2023, Lin et al., 2022), and are therefore of greater relevance in the context of the Executive Order.
More information about biological sequence data can be found in Appendix A, and our data collection methodology can be found in Appendix B.
Training compute
Over the past 6 years, the amount of computing power used to train biological sequence models has grown swiftly. We find that, on average, the compute budgets for training these models have increased by 8-10 times per year (see Appendix C). This rate of growth in compute usage is faster than what has been seen for computer vision models and reinforcement learning systems during the same time period. However, it is comparable to the growth rate in compute for language models between 2018 and 2024 (Epoch AI, 2024). Frontier biological sequence models today, such as ESM2-15B (Lin et al., 2022), ProtBERT, ProtT5-XXL (Elnaggar et al. 2021), and the Nucleotide Transformer (Dalla-Torre, 2023) are trained on 1e22 FLOP or more by a range of academic and industry labs.
Notable is xTrimoPGLM-100B (Chen et al., 2023), a 100B parameter protein language model trained on 1T protein tokens by researchers from Tsinghua University and BioMap Research, a Chinese-based biotech company. Trained on 768 NVIDIA A100s between January 18 and June 30, 2023, the model took around 6e23 operations to train, well over the 1e23 threshold set by the Executive Order.3 xTrimoPGLM-100B outperforms smaller models such as Meta’s ESM2-15B on most benchmark tasks, and shows strong potential in creating novel protein sequences with valid 3D structures (ibid.)

Figure 1. Scatter plot depicting the evolution of training compute requirements (in FLOP) for 46 systems over time, from 2018 to 2023. Each point represents a system, labeled with its name, plotted against its publication date and the logarithm of its training compute.
While the compute used to train xTrimoPGLM-100B is a few-fold larger than models such as ESM2-15B or ProtBERT, it is relatively small by the standards of language modeling. For example, xTrimoPGLM-100B is roughly at the scale of GPT-3 (Brown et al., 2020), a model released three calendar years earlier. Top industry labs leading on protein sequence models, such as Meta and Google DeepMind, regularly train models with ten or a hundred fold more resources for other purposes. Unlike leading large language models which are often proprietary and closed source, the vast majority of biological sequence models in our dataset make their code and weights freely available for download.
Large language models trained on biological data
Most of the models in our dataset are trained exclusively on biological sequence data. However, some are large language models that are trained primarily on text corpuses, and only partially trained or fine-tuned on biological data. Notable among these is the language model Galactica-120B (Taylor et al., 2022), which was trained on over 1e23 operations. Its training data include biological datasets like UniProt and RefSeq. However, since biological data did not constitute a majority of its training data, Galactica-120B likely does not fall under the Executive Order’s additional reporting requirements. Llama-molinst-protein-7b (Fang et al., 2023) is another example of a language model partially trained on biological data. It is a fine-tuned LLaMA-7b model, trained on the Mol-Instructions dataset, which contains protein-oriented instructions for predicting protein structure, function, and activity, with the aim of facilitating protein design based on textual directives. Although Llama-molinst-protein-7b has capabilities for biological sequence modeling, like Galactica it was not primarily trained on biological sequence data.
In one interpretation of the Executive Order, this represents a potential gap in the reporting requirements of the recent Executive Order. Models trained on less than 1e26 FLOPs could potentially incorporate all known protein sequences while evading oversight by not being primarily biological. For example, a foundation model trained using 1e25 FLOP typically trains on around ten trillions of tokens (Hoffmann et al., 2022). Such a model could potentially include the entire known corpus of around 7 billion protein sequences within its dataset, and not be subject to additional reporting requirements due to the biological sequence data being a minority of its training data.4 Yet such models may match or exceed the capabilities of smaller, exclusively biological models that do meet the bar for additional governance requirements.
Biological sequence data
The Executive Order highlights the national security implications of biological datasets, particularly ones related to pathogens and omics studies.5 The order directs agencies to conduct detailed security reviews of their data assets.6 In this section, we examine the availability, generation, and usage of biological sequence data.
Biological sequence data used to train biological sequence models is provided by a vast array of public databases compiled by government, academic, and private institutions. We delineate major sources into three primary categories:
-
DNA sequence databases. These have the highest growth rate of analyzed databases, with GenBank seeing a 31% increase in the number of sequences stored between 2022 and 2023. Whole genome shotgun sequencing7 studies have been the driving force of growth of DNA data, as the increase in number of entries in all other GenBank divisions, referred to as traditional entries, is greatly attenuated in comparison.
-
Protein sequence databases. The level of detail in protein sequence databases can vary. Databases with rich annotations such as UniProtKB have a much slower growth rate (6.7%), compared to metagenomic databases such as MGnify (20%), which provide protein sequences but lack detailed information about the protein’s structure, function, and origin.
-
Protein structure databases. Gathering experimental data on protein structures is slow and painstaking. Thus, the Protein Data Bank grows by only 6.5% per year. Instead, databases publishing protein structures predicted by AI models can quickly generate large volumes of synthetic data. Databases of synthetic data such as AlphaFoldDB and ESMAtlas have dramatically boosted the supply of available data, though their growth could slow as opportunities for synthetic data are exhausted.
The majority of entries in large biological databases such as the International Nucleotide Sequence Database Collaboration (INSDC), MGnify, UniProtKB and PDB pertain to cellular organisms (humans, animals, plants, fungi, yeast, bacteria). For example, UniProtKB entries comprise 97% cellular and 2% viral protein sequences, a subset of which are known pathogens.
| Data type | Key datasets | Number of entries | Provider/main funder | Growth rate 2022-2023 | Method of data generation | Key models trained on this type of data |
|---|---|---|---|---|---|---|
| DNA sequences | INSDC (GenBank, ENA, DDBJ) | 3.4B | NCBI (US), ENA (Europe), DBBJ (Japan) | 31.3% [GenBank] | DNA sequencing (Next-generation and Sanger sequencing) | CaLM, Nucleotide Transformer, HyenaDNA |
| Protein sequences | MGnify protein database | 3B | EMBL-EBI (Europe) | 20.0% [MGnify] 15.21% [UniRef50] 6.7% [UniProtKB] 0.39% [UniProtKB/ Swiss-Prot] 6.75% [UniProtKB/ TrEMBL] | Majority of protein sequences are inferred from known DNA/RNA sequences | ESM2, ESM1b, ESM1v, ProtT5, ProtBert, ProGen, RITA, Ankh, gLM, xTrimoPGLM, AlphaFold, AlphaFold 2 |
| BFD | 2.5B | Steinegger M. and Söding J. (Academics) | ||||
| MetaClust | 1.6B | Steinegger M. and Söding J. (Academics) | ||||
| UniParc | 596M | PIR (US), SIB (Swiss), EMBL-EBI (Europe) | ||||
| UniRef | 365M | PIR (US), SIB (Swiss), EMBL-EBI (Europe) | ||||
| UniProtKB | 252M | PIR (US), SIB (Swiss), EMBL-EBI (Europe) | ||||
| DNA sequences linked to RNA and protein sequence | RefSeq | 411M | NCBI (US) | 13.2% [RefSeq] | ||
| Protein structures | ESMAtlas | 772M | Meta | 6.5% [PDB] | AI predicted | AlphaFold, AlphaFold 2 |
| AlphaFoldDB | 214M | Google DeepMind | ||||
| PDB | 213k | Worldwide PDB (Global) | Experimentally derived (X-ray, NMR, cryo-EM) | |||
| SCOPe | 107k | CPE and LMB (UK) |
Table 1. Overview of key data sources for biological sequence models. Number of entries refers to either the number of DNA sequences, protein sequences, clusters of protein sequences, protein structures or clusters of protein structures depending on the database. NMR refers to Nuclear Magnetic Resonance (NMR) spectroscopy, and cryo-EM Cryogenic electron microscopy, two techniques for studying protein structures.
While protein sequence data is abundant, detailed functional knowledge is limited to a smaller subset, such as those in UniProtKB-Swiss-Prot. These are either manually annotated (UniProtKB-Swiss-Prot) or automatically annotated (UniProtKB-TrEMBL) with ligand binding sites, subcellular localizations, 3D structures and known functions. All UniRef datasets are derived from sequences stored in UniProtKB and UniParc, and represent the most commonly used training datasets for models analyzed in this report (Supplementary Figure 3).
One important source of genetic data is metagenomic sequencing. Metagenomic sequencing is the practice of sequencing genomic data (DNA/RNA) of multiple organisms. Specifically, organisms such as bacteria, within samples from diverse environments (eg. soil, ocean, microbiomes). Metagenomic databases include MGnify, MetaClust and BFD (combination of MetaClust and UniProtKB curated for training AlphaFold). Protein sequences in these metagenomic databases are inferred from the sequenced coding regions in DNA. The majority of entries lack either the N- or C-terminus of the sequence, as many genes sequenced in metagenomic studies are truncated. Additionally, protein sequences derived from metagenomic databases commonly lack detailed functional annotation.
Metagenomic data is commonly included in pre-training stages to diversify datasets. For example, supplementing high quality UniProtKB-based datasets with vast amounts of metagenomic data improved AlphaFold’s performance at CASP14 (Jumper et al. 2021). Likewise, xTrimoPGLM-100b’s training regime included pre-training with 1.1B sequences from metagenomic databases (BFD, MGnify, MetaClust) in addition to UniRef90, prior to training with a redundancy-refined subset emphasizing higher-quality UniRef90 data (Chen et al. 2023). Thus, despite its lack of detailed annotations, data from metagenomic sequencing is a key data source for biological sequence models.
Trends in biological sequence data
Biological sequence data has accumulated steadily over the past forty years. We observe a rapid but sub-exponential growth in the number of entries across the main public biological sequence databases, including DNA, protein sequence, and protein structure databases. We observe a large increase in the number of biological sequence databases around the turn of the century, with the emergence of multiple UniProt databases such as UniProtKB, UniRef, and UniParc, as well as MGnify and Metaclust. Later, the emergence of AI-predicted databases, notably AlphaFoldDB and the ESM Metagenomic Atlas exhibited a rapid growth trajectory, underscoring the impact of machine learning techniques in the generation of protein structure data.

Figure 2. Growth of key biological sequence databases between January 1976 and January 2024.
Number of entries corresponds to number of DNA sequences in GenBank, ENA, DDBJ, number of records in RefSeq, number of protein sequences in MGnify, UniParc, UniRef, UniProtKB and number of structures in PDB, AlphaFoldDB and ESMAtlas.
To improve performance on downstream tasks, biological sequence models benefit from training on large quantities of biological sequence data. But different databases often contain duplicates of individual entries. Therefore AI developers typically aggregate multiple datasets and then refine the data by removing duplicates. To approximate the order of magnitude of currently available protein sequence data for training, and therefore give an intuition for potential for scaling biological sequence models, we estimated the unique entries in key public protein sequence databases. As databases have substantial overlap, the number of entries cannot simply be summed. Instead, we produced a rough estimate by summing over non-overlapping dataset components. Specifically, UniParc (596 million), MGnify (3 billion), the soil reference catalog within BFD (2 billion), the marine reference catalog within BFD (292 million), and MetaClust (1.59 billion) summate to approximately 7.5 billion sequences.8 If the smaller UniProtKB (252 million, a subset of UniParc) were included instead, the total would be around 7.1 billion unique sequences.
While imprecise, this compilation gives a sense of the scale of accumulated protein sequence data, which exceeds 7 billion entries across these key repositories. Moreover, in practice, datasets are often clustered according to different sequence similarity thresholds prior to training to reduce the amount of sequences with high similarity to each other in the dataset. Nevertheless, this estimate suggests that the current stock of data is likely to be nearly a factor of ten greater than what has been used in the largest training runs so far.
Sources of biological sequence data
There are three main data generation methods: DNA sequencing, inference from known DNA/RNA sequences, and experimental protein structure determination. DNA sequencing is used for generating DNA sequence databases such as those found in the INSDC, which includes GenBank, ENA, and DDBJ. Prominent sequencing technologies used include Next-generation sequencing (NGS) (commercially available since 2005) and traditional Sanger sequencing (commercially available since 1986).
The massively parallelized NGS sequencing enabled by next generation sequencing technologies commercially offered by Illumina, Oxford Nanopore and Pacific Biosciences, have been instrumental in compiling the 3.4 billion entries in the DNA sequences databases of the INSDC (Hershberg, 2022).This trend is mirrored by the sequence read archive (SRA), which is part of the INSDC collaboration, and contains exclusively raw sequence reads produced by NGS technologies. Many sequences in the SRA are associated with entries in GenBank/ENA/DDBJ and other large-scale sequencing projects of human genomes such as the 1000 Genomes project, which have relied on NGS technologies. The amount of storage space required for data in SRA has grown exponentially in the last decade, increasing by roughly 40% between 2013 and 2019 (NCBI Insights, 2020).
The majority of sequences in protein sequence databases like MGnify, MetaClust and UniProtKB are derived from the translation of known coding DNA/RNA sequences. This approach has been applied to amass billions of protein sequences, with over 95% of the 252 million sequences in UniProtKB being derived from DNA sequences in INSDC and MGnify alone housing 3 billion sequences.
Finally, experimental methods are commonly used to generate structure data. Protein structures in databases such as PDB are acquired through experimental methods like X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy. Additionally, AI-prediction methods contribute to the growth of structural databases, as seen with the 772 million entries in ESMAtlas and 214 million entries in AlphaFoldDB.
Discussion
The recently issued White House Executive Order establishes comprehensive new reporting and oversight requirements for AI models trained on biological sequence data that could present dual-use risks. Biological sequence models exceeding 1e23 operations (or 1e26 for all models) will face mandatory disclosures on model details, red team evaluations probing misuse potential, cybersecurity protections, and controls around access to model weights. However, further regulatory clarification may be needed, as models mixing biological data while under the 1e26 operations threshold could plausibly match or exceed the capabilities of smaller pure biological sequence models yet evade governance measures.
The analysis reveals xTrimoPGLM-100B exceeds the 1e23 operations threshold by a factor of at least 6x. We find that training compute is increasing rapidly, even at the standards of the field of machine learning more broadly, with biological models seeing budgets grow 8-10x yearly. This growth in compute is consistent with broader trends in machine learning where enhanced computational capabilities and data availability are driving rapid advancements.
The Executive Order underscores the necessity of addressing security concerns related to biological sequence datasets used in training models, particularly those containing pathogenic information. Our survey has identified a vast and rapidly expanding array of biological data across DNA, protein, and structure databases facilitated by advancements in sequencing technologies and AI prediction methods.
From a policy and regulatory standpoint, our findings highlight potential gaps in the current regulatory frameworks governing AI models that handle biological data. The Executive Order’s emphasis on models trained on “primarily biological sequence data” may inadvertently allow models that incorporate significant but not majority biological data to evade stringent oversight, despite using more than 1e23 operations during training. This could potentially result in models with extensive biological sequences not being subject to the heightened governance intended for high-risk biological sequence models. There is a need for policymakers and regulatory agencies to consider how the current frameworks might need adaptation to keep pace with the new approaches to AI training, such as large-scale multi-domain and multi-modal training.
Acknowledgments
We thank Oliver Crook, Simon Grimm, Adam Marblestone, Ben Cottier, David Owen, Matthijs Maas, Juan Cambeiro, Jaime Sevilla, Misha Yagudin, and Martin Stoffel for their helpful comments, and Edu Roldán for his support with uploading and formatting our work on the website.
Appendix A: Biological sequence background information
The central dogma outlines the flow of genetic information from DNA to RNA to proteins. This framework describes how genes are transcribed into RNA, which is then translated into amino acid sequences that fold into functional proteins. This appendix offers a short overview of how these key processes of transcription, translation, and protein folding convert genes to functional proteins.
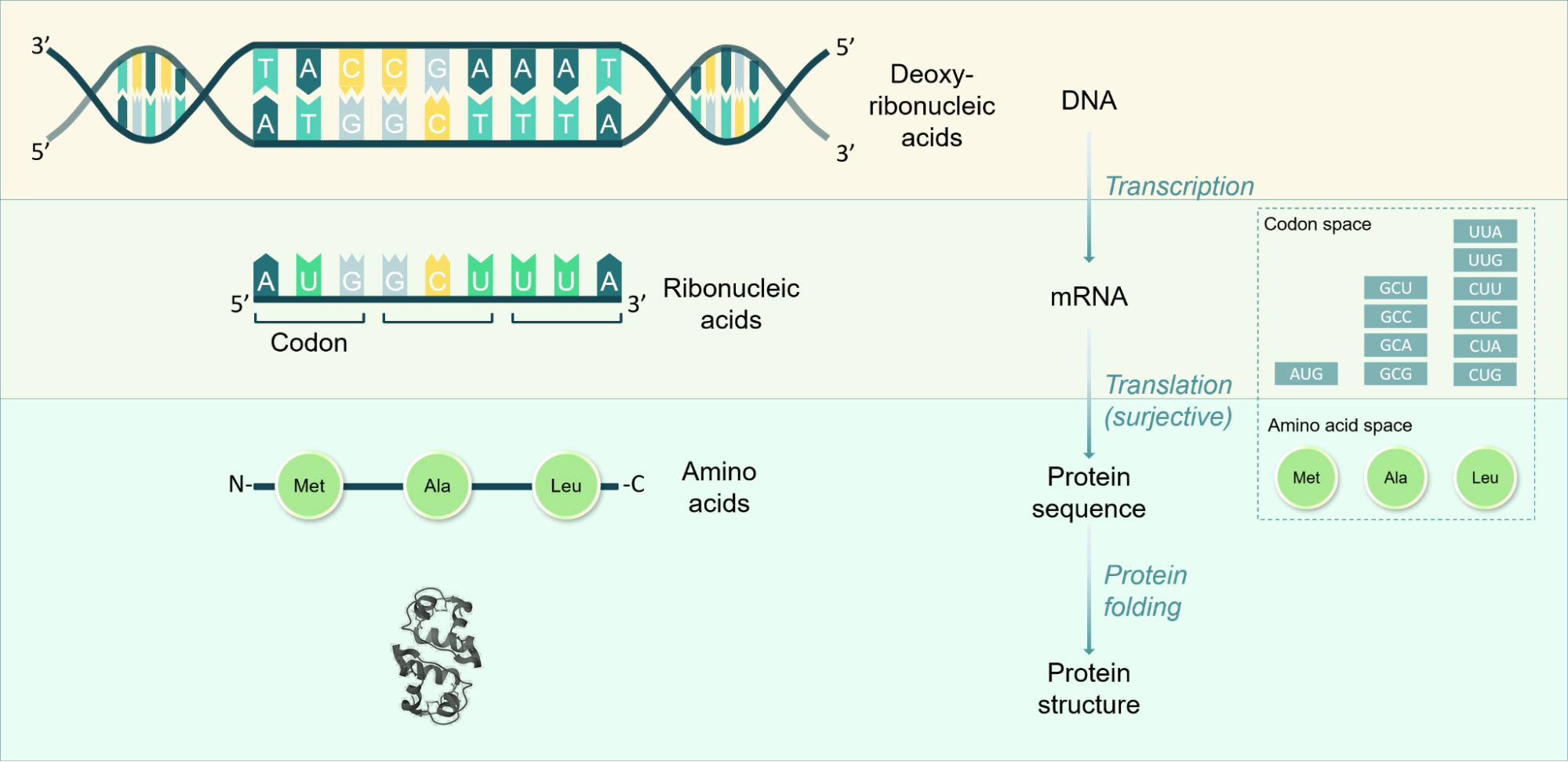
Supplementary Figure 1. Overview of the central dogma of biology, which describes the relationships between the different types of biological sequence data discussed in this report. Example structure shown is human insulin precursor dimer from PDB.
Transcription: DNA molecules are double-stranded sequences consisting of pairs of bases of complementary deoxyribonucleic acids. Coding regions of DNA contain sequences of base pairs commonly referred to as genes, which can be translated to synthesize proteins as opposed to non-coding regions which contain sequences that regulate in what circumstances coding regions are transcribed. Transcription refers to the process in which DNA is transcribed to a single-stranded sequence of ribonucleic acids, whereby uridine (U) is integrated as the complementary base to adenosine (A) instead of thymidine (T). There are 4 nucleotides commonly found in DNA (adenine (A), thymine (T), cytosine (C), and guanine (G)) and 4 commonly found in RNA (adenine (A), uracil (U), cytosine (C), and guanine (G)).
Translation: Single stranded messenger RNA (mRNA) sequences are the bases of the process of translation, in which ribosomes recognize triplets of RNA bases, referred to as codons, and add a specific corresponding amino acid to a growing amino acid sequence chain. Translation is a surjective process, in that a given codon codes for a single amino acid, but multiple codons can code for the same amino acid. Therefore, amino acid sequences can be inferred from DNA and RNA sequences, but there is no unique RNA/DNA sequence that can be inferred from the amino acid sequence. There are 20 common amino acids that form the building blocks of the majority of all proteins.
Protein folding: Protein sequences fold into their 3D structure spontaneously and are then able to perform their functions. This spontaneous process tends to occur on the time scale of milliseconds and is a non-trivial process to predict, given the many possible theoretical conformations a protein may take on and the many interactions between distant amino acids in the sequence once folded. Experimentally, X-ray crystallography is an important method for structure verification and involves integrating the protein into a crystal lattice prior to subjecting it to X-ray beams to determine diffraction patterns. Similarly, a newer method is cryogenic electron microscopy (cryoEM), in which proteins are placed in a water-based solution that is frozen on a short enough timescale to form non-crystalline ice, to allow imaging with an electron microscope to determine high-resolution protein structures.
Biological sequences compared to natural language: Biological sequences consist of a much smaller vocabulary than natural languages, with a handful of nucleic acids (and 64 possible nucleic acid triplets) and 20 amino acids forming the basic characters of biological sequences. However, biological sequences tend to be hundreds of characters long and contain long-range dependencies compared to natural language sentences, which in the case of English have on average 15-20 words.
Appendix B: Data collection methodology
Our dataset was built based on surveying the biological sequence model literature, leaderboards of common benchmarks used to evaluate models and frequently downloaded open-source protein language models. Our key sources included:
| Source | Description |
|---|---|
| Hugging Face | Included popular public open-source protein language models |
| ProteinGym (Notin et al. 2023) | Included models reported in ProteinGym benchmark publication which evaluated models on a collection of benchmarks comparing the ability of models to predict the effect of protein mutations on protein fitness and other metrics. |
| PEER Integrated Leaderboard | Included subset of models in the leaderboard covered in PEER benchmark for protein sequence understanding, which is a comprehensive multi-task benchmark |
| Functional predictions benchmark (Unsal et al. 2022) | Included models covered in Table 1, which used deep learning methods |
| Surveys of protein language models (e.g. Hu et al., 2022) | Included models from Table 1 of Hu et al. 2022, which conducted an extensive survey of pre-trained protein language models |
| Models submitted to CASP12 and CASP13 | Included notable models, which competed in CASP12-CASP13 to give a greater historic context of models used to predict protein structures prior to AlphaFold 2’s breakthrough success in CASP14 |
Supplementary Table 1. Key data sources for building dataset of biological sequence models
Our database growth data was collected from published statistics on the database (if available) or extracted from release notes published with every database release and is detailed in Supplementary Table 2.
| Database | Data source | Code to extract from release notes |
|---|---|---|
| GenBank-traditional | Extracted from release notes | Notebook 1 |
| GenBank-all | ||
| ENA | Based on release note Appendix A “Database growth table” | |
| DDBJ | Based on the table in the most recent release note 10. Release History | Notebook 2 |
| RefSeq | Based on the Release Note Section 3.9 “Growth of RefSeq” | |
| MGnify | Extracted from release notes | Notebook 3 |
| UniParc | Data 2014-2023 extracted from release notes Data 2004-2014 released in Supplementary Material of UniProt 2015 paper | Notebook 4 |
| UniRef100 | ||
| UniRef90 | ||
| UniProtKB | ||
| UniProtKB/Swiss-Prot | ||
| UniProtKB/TrEMBL | ||
| PDB | CSV downloaded from PDB statistics | |
| AlphaFoldDB | 4 different releases from July 2021-July 2022 tracked in Figure 1 in Varadi et al. 2024 | |
| ESMAtlas | Datapoints based on ESMAtlas website |
Supplementary Table 2: data sources for database growth statistics
To estimate compute used during training, we use the methodology described by Sevilla et al., 2022.
Appendix C: Growth in training compute
Our dataset includes models trained on low amounts of compute and trained with much larger compute budgets. As such, the variance is high, and a single exponential trend fits the data poorly. Hence, we filter out the lowest-compute budget training runs, and estimate a simple exponential function. Since there is no principled way to decide a cutoff, we apply a range of filters (filtering out the bottom 20th, 30th, and 40th percentile of models within each contiguous 365-day period), and estimating the growth rate using the filtered data.



Supplementary Figure 2: Growth in compute budgets used to train biological sequence models across three subsets of the data.
We find growth rates of 8.7x to 10.4x/year (0.94 to 1.02 orders of magnitude/year), see Supplementary Figure 2. This is notably faster than growth in other domains of AI, such as vision or language modeling (Epoch AI, 2023).
Appendix D: Use of biological sequence datasets
This appendix details the use of various biological sequence datasets for training models within our database. Supplementary Figure 3 depicts the number of models trained on each dataset, specifically filtered to display datasets used by more than one model.

Supplementary Figure 3. Number of models trained on each dataset in our database. Filtered to only show datasets used by >1 model.
UniRef50 is the most commonly used dataset, with 23 models trained, followed by UniRef90 with 11 models trained. PDB is the most commonly used structure database, and is employed in the training of 6 models.
-
The scaling of frontier models over the same period has been roughly 5x/year (Epoch AI, 2024), for vision models it is around 4x/year (Epoch AI, 2024), while for language modeling the amount of compute used has grown faster, by around 10x/year (Epoch AI, 2024).

-
Google DeepMind (then DeepMind) spun out Isomorphic Labs in 2021 to build on AlphaFold 2’s breakthrough in the field of drug discovery and have announced that new iterations of AlphaFold will significantly improve predictions of joint structures of complexes involving proteins, nucleic acids, small molecules, ions and modified residues. Such predictions for biomolecular interactions are likely to be especially important for understanding protein function.
-
In fact, the model might be trained longer than this. The latest preprint (as of July 6, 2023) states that: “As of the current date, xTrimoPGLM-100B continues its pre-training process in a unified manner, with the aim of further enhancing its modeling power.”
-
The current corpus of known biological sequence data consists of approximately 7 billion protein sequences as detailed in the Biological Sequence Data section. With an average length of 200 amino acids per sequence and tokenizing to 1 token per amino acid, the total representable biological sequence dataset equates to roughly 1.4 trillion tokens. Today’s state-of-the-art LLMs such as GPT-4, Gemini, and others are typically trained using on the order of 10T tokens. Hence, the biological sequence data would constitute a minority of the total training data, if biological sequence data was folded in.
-
The Executive Order considers “the national security implications of the use of data and datasets, especially those associated with pathogens and omics studies, that the United States Government hosts, generates, funds the creation of, or otherwise owns, for the training of generative AI models, and makes recommendations on how to mitigate the risks related to the use of these data and datasets” (Section 4.4, part ii, B).
-
The order mandates that “within 180 days of the development of the initial guidelines required by subsection 4.7(a) of this section, agencies shall conduct a security review of all data assets in the comprehensive data inventory required under 44 U.S.C. 3511(a)(1) and (2)(B) and shall take steps, as appropriate and consistent with applicable law, to address the highest-priority potential security risks that releasing that data could raise with respect to CBRN weapons, such as the ways in which that data could be used to train AI systems” (Section 4.7, part b).
-
Entails randomly fragmenting DNA strands and then sequencing the many overlapping DNA fragments in parallel. Once sequenced, overlapping fragments are aligned and assembled in silico to obtain whole genome sequences.
-
We sum over only the soil and marine reference catalogs within the BFD to avoid double counting, given that the BFD includes UniProtKB, and MetaClust.
Updates
About the authors



Related work